
Imaginez un copiste médiéval chargé de reproduire un manuscrit. Pendant des heures, sa plume glisse sans accroc sur le parchemin — jusqu’à ce qu’elle tombe sur cette phrase : « et ainsi ainsi ainsi ainsi ainsi le roi décida… » Ses yeux accrochent. Il en saute une, ou en double une. Quand il se relève, il ne sait plus s’il en était à la troisième ou la quatrième répétition. La faute est minuscule. Elle est aussi, d’une certaine façon, inévitable.
Notre ADN vit cette scène des millions de fois par jour. Et ce qui ressemble à une défaillance pourrait bien être, sous un autre angle, l’une des plus ingénieuses archives biologiques que l’évolution ait produites.
Le génome humain est troué de zones étranges : des motifs courts — quelques bases azotées, par exemple un dinucléotide comme CA répété vingt fois de suite — qui se répètent en tandem des dizaines, parfois des centaines de fois. Ces répétitions en tandem courtes (microsatellites, connus en anglais sous l’acronyme STR pour short tandem repeats) ne codent aucune protéine. Elles ne semblent exprimer rien d’utile. Pendant des décennies, on les a parquées sans cérémonie dans la catégorie de l’ADN « poubelle » — terme révélateur, qui en dit peut-être plus sur les certitudes d’une époque que sur la réalité du génome.
Les microsatellites ont pourtant trouvé leur utilité bien avant d’intéresser les biologistes de l’évolution. En médecine légale, leurs profils de longueur servent depuis les années 1990 à identifier des individus avec une précision remarquable. En génétique des populations, ils permettent de retracer des migrations et des brassages génétiques sur quelques générations. C’est de cette double réputation — marqueur instable mais informatif — qu’une prépublication récente tire tout son intérêt.
Mais les zones silencieuses ont souvent des histoires à raconter, à condition de savoir les écouter.
Lors de la réplication, l’ADN polymérase — l’enzyme qui duplique notre patrimoine génétique à chaque division cellulaire — bute sur ces séquences monotones. Elle glisse. Elle ajoute une répétition de trop ou en efface une. Les généticiens ont un mot pour cela : bégaiement (en anglais stutter). La métaphore est juste. Sur ces tronçons du génome, la machinerie moléculaire trébuche, comme une langue qui accroche sur une syllabe difficile.
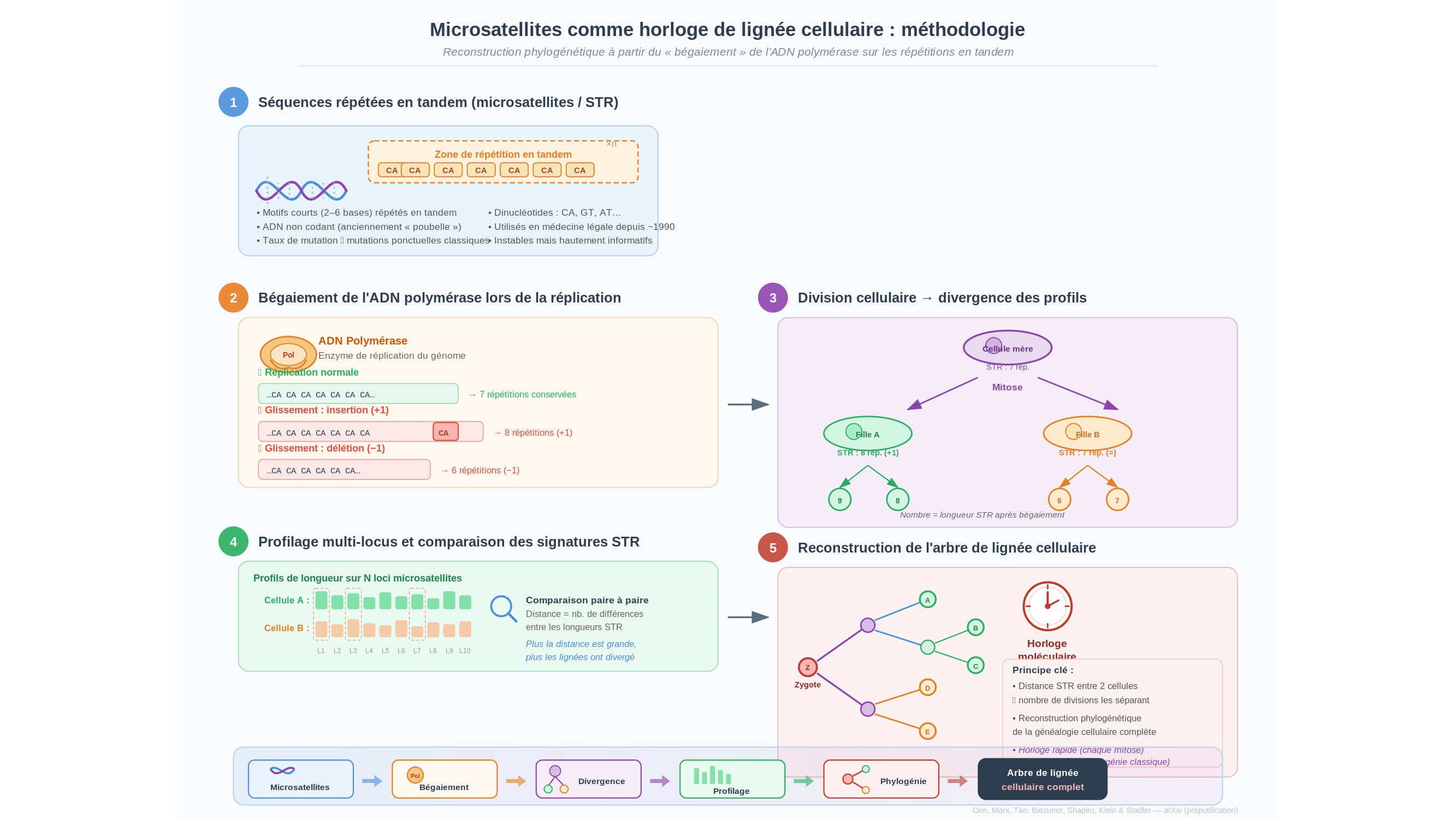
Ce bégaiement est documenté depuis plusieurs décennies. La littérature sur les microsatellites établit que leur taux de mutation — la probabilité qu’une répétition change de longueur lors d’une division cellulaire — dépasse de plusieurs ordres de grandeur le taux habituel de mutation ponctuelle du génome. Ce n’est pas la même temporalité. C’est un monde à part, où l’instabilité est la règle et la constance, l’exception. [Note de la rédaction : les chiffres précis varient selon les études et les types de motifs ; une référence de synthèse sera ajoutée avant publication.]
Ici surgit le paradoxe qui est au cœur d’une prépublication récente déposée sur arXiv. Amos Onn, Tzipy Marx, Liming Tao, Tamir Biezuner, Ehud Shapiro, Christoph Klein et Peter Stadler — sept chercheurs affiliés à l’Université de Regensburg, à l’Institut Weizmann, à l’Université de Leipzig et à plusieurs partenaires — ont retourné ce défaut comme un gant. L’instabilité des microsatellites, loin d’être du bruit moléculaire, serait une horloge. Pas l’horloge lente et régulière de la phylogénie classique, mais une horloge rapide, cellule après cellule, dont les tics correspondent aux divisions qui séparent deux lignées.
L’idée est élégante : chaque fois qu’une cellule se divise, elle transmet à ses filles une longueur légèrement différente de ses microsatellites. Ces longueurs s’accumulent, divergent, forment peu à peu une signature propre à chaque lignée. En comparant les profils de microsatellites de deux cellules, on peut — selon toute vraisemblance en recourant à un formalisme mathématique de type chaîne de Markov en temps continu, même si les détails précis du modèle méritent d’être vérifiés dans le texte intégral — estimer le nombre de divisions qui les séparent. Sans séquencer l’intégralité du génome. Les bégaiements suffisent.
L’application la plus immédiate, et sans doute la plus pressante, concerne le cancer. Une tumeur est, en miniature, une évolution somatique : une lignée cellulaire qui diverge, accumule des variations, sélectionne les clones les plus agressifs. Reconstituer l’arbre de cette divergence — identifier quelles cellules cancéreuses descendent de quelles autres, à quel moment une bifurcation décisive s’est produite — c’est comprendre comment la maladie s’est construite, et peut-être comment l’interrompre. Les microsatellites, avec leur mutation rapide et régulière, offrent ici une résolution temporelle que les méthodes classiques de séquençage ne permettent pas d’atteindre aussi facilement.
La même logique s’applique à l’embryogenèse : retracer comment un œuf unique donne naissance à des milliards de cellules différenciées, reconstituer l’arbre généalogique des tissus, mesurer les dérapages qui surviennent tôt dans le développement. Le périmètre de l’étude est celui des lignées cellulaires au sein d’un organisme, ou de populations récentes — et non la grande phylogénie des espèces.
Il faut pourtant regarder cette promesse avec un peu de recul. Plusieurs limites méritent d’être nommées.
La première est technique. Les microsatellites sont notoirement difficiles à séquencer avec précision : les technologies de séquençage courte lecture produisent fréquemment des erreurs sur ces zones répétitives, et distinguer une vraie variation biologique d’un artefact expérimental reste un défi actif du domaine. Un modèle aussi élégant que celui d’Onn et ses collègues ne vaut que si les données en entrée sont fiables — ce qui n’est pas toujours garanti dans les tissus tumoraux, où la qualité de l’ADN est souvent dégradée.
La seconde est plus fondamentale. Les microsatellites ne mutent pas tous au même rythme. Leur taux de variation dépend du motif répété, de la longueur de la répétition, du contexte génomique environnant — et probablement d’autres facteurs encore mal compris. Calibrer une horloge suppose que l’on connaisse son tic-tac. Or la prépublication modélise ces dynamiques mutationnelles avec un niveau de précision qui reste à évaluer dans des contextes biologiques variés.
Il y a quelque chose de légèrement vertigineux dans tout cela. Pendant longtemps, la biologie a cherché l’information génétique dans ce qui était stable, conservé, protégé de l’erreur. Les gènes fonctionnels, les séquences régulatrices, les régions que la sélection naturelle semblait tenir à l’abri des mutations. Et voilà que l’on trouve de l’information précisément dans les zones les plus instables, celles que l’on avait cru dispensables.
L’histoire des bégaiements du génome ressemble un peu à celle des cicatrices : on pensait qu’elles ne disaient rien, et c’est là qu’elles gardent le mieux la mémoire de ce qui s’est passé.
Sources
Amos Onn, Tzipy Marx, Liming Tao, Tamir Biezuner, Ehud Shapiro, Christoph A. Klein, Peter F. Stadler — Modeling the mutational dynamics of very short tandem repeats — arXiv:2603.25628 — https://arxiv.org/abs/2603.25628
[Référence secondaire à ajouter avant publication : revue de synthèse sur les microsatellites comme marqueurs phylogénétiques — arXiv ID ou DOI requis.]


