L’empreinte de la destruction : lire l’orientation d’une protéine dans ses débris
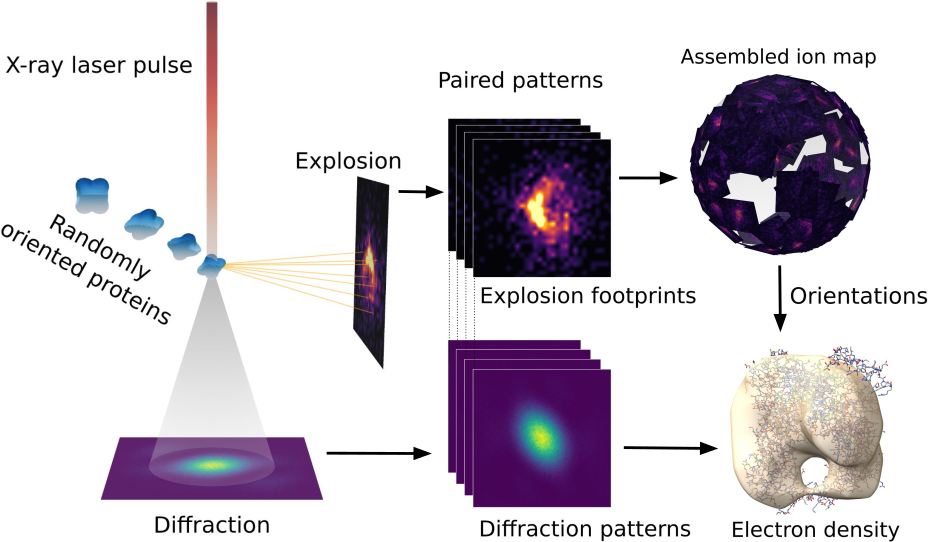
Il y a quelque chose de vertigineux dans l’idée que pour voir une molécule, il faut d’abord la détruire. Pas l’abîmer, pas l’effleurer — la pulvériser. En quelques femtosecondes, un laser d’une puissance inouïe arrache chaque électron de la protéine, laissant les noyaux atomiques nus, chargés positivement, se repousser mutuellement avec une violence que rien ne peut contenir. La molécule explose. Et c’est précisément dans cette explosion que Tobias André, Alessandro Bellisario, Nicusor Timneanu, Carl Caleman et leurs collègues de l’université d’Uppsala ont décidé de chercher une information que personne n’avait songé à y lire : l’orientation de la protéine au moment du tir.
Pour comprendre pourquoi cette question est si cruciale, il faut d’abord saisir le défi que représente l’imagerie des protéines en phase gazeuse — c’est-à-dire isolées, molécule par molécule, dans le vide. C’est la promesse des lasers à électrons libres de type XFEL (X-ray Free Electron Laser), des installations monumentales — plusieurs kilomètres de long, des centaines de millions d’euros d’investissement — capables de tirer des impulsions X d’une brièveté et d’une intensité sans équivalent dans la nature. L’idée est séduisante : photographier une protéine seule, sans la contraindre dans un cristal, sans l’artefact des conditions de cristallisation. Voir la molécule telle qu’elle existe vraiment.
Mais il y a un obstacle de taille. En phase gazeuse, chaque protéine tourne librement sur elle-même. Quand le laser déclenche, il fige la molécule dans une pose inconnue. Imaginez mille photographies d’une même sculpture, prises dans le noir complet, sans jamais savoir sous quel angle l’objectif était pointé. Comment, à partir de ce chaos d’images désalignées, reconstruire la forme en trois dimensions ?
La méthode classique consiste à exploiter les figures de diffraction produites par les rayons X eux-mêmes. C’est élégant en théorie, mais redoutablement difficile en pratique : avec une seule molécule, le signal est si faible qu’il faut accumuler des centaines de milliers de tirs, et l’algorithme de reconstruction dit EMC (Expansion-Maximisation-Compression) — qui tente d’inférer l’orientation de chaque cliché par recoupements successifs — peine encore à converger. La montagne de données nécessaires est décourageante.
L’équipe d’Uppsala a retourné le problème. Et si l’explosion, que tout le monde considère comme un dégât collatéral inévitable, n’était pas une perte mais une ressource ? Les ions éjectés lors d’une explosion de Coulomb ne partent pas dans n’importe quelle direction : leurs trajectoires dépendent de la géométrie initiale de la molécule. Chaque atome s’échappe en suivant la ligne de force du champ électrique que lui imposent ses voisins — et cette ligne de force est dictée par la structure tridimensionnelle de la protéine. L’explosion laisse donc une empreinte spatiale, aussi singulière qu’une empreinte digitale.
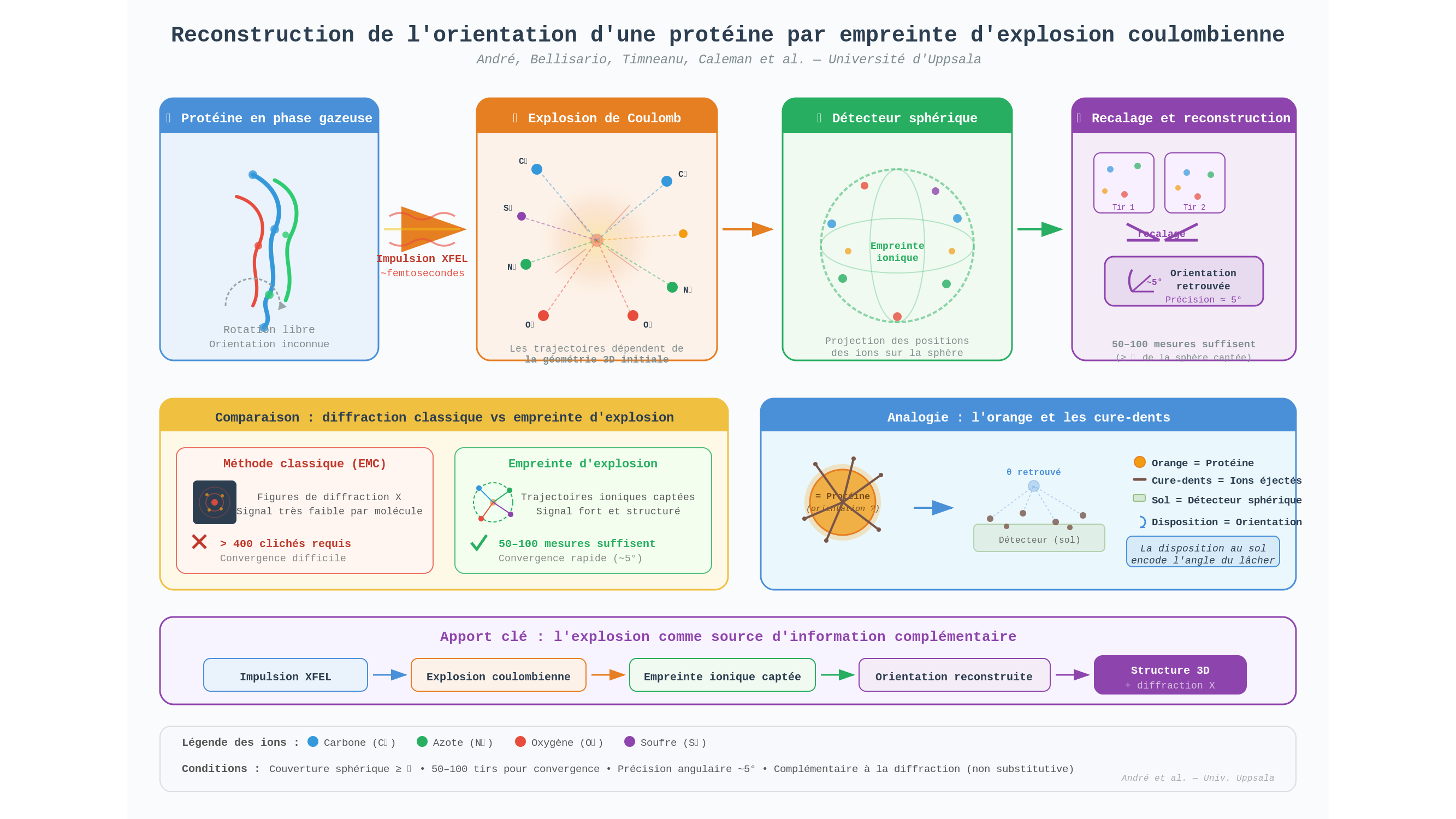
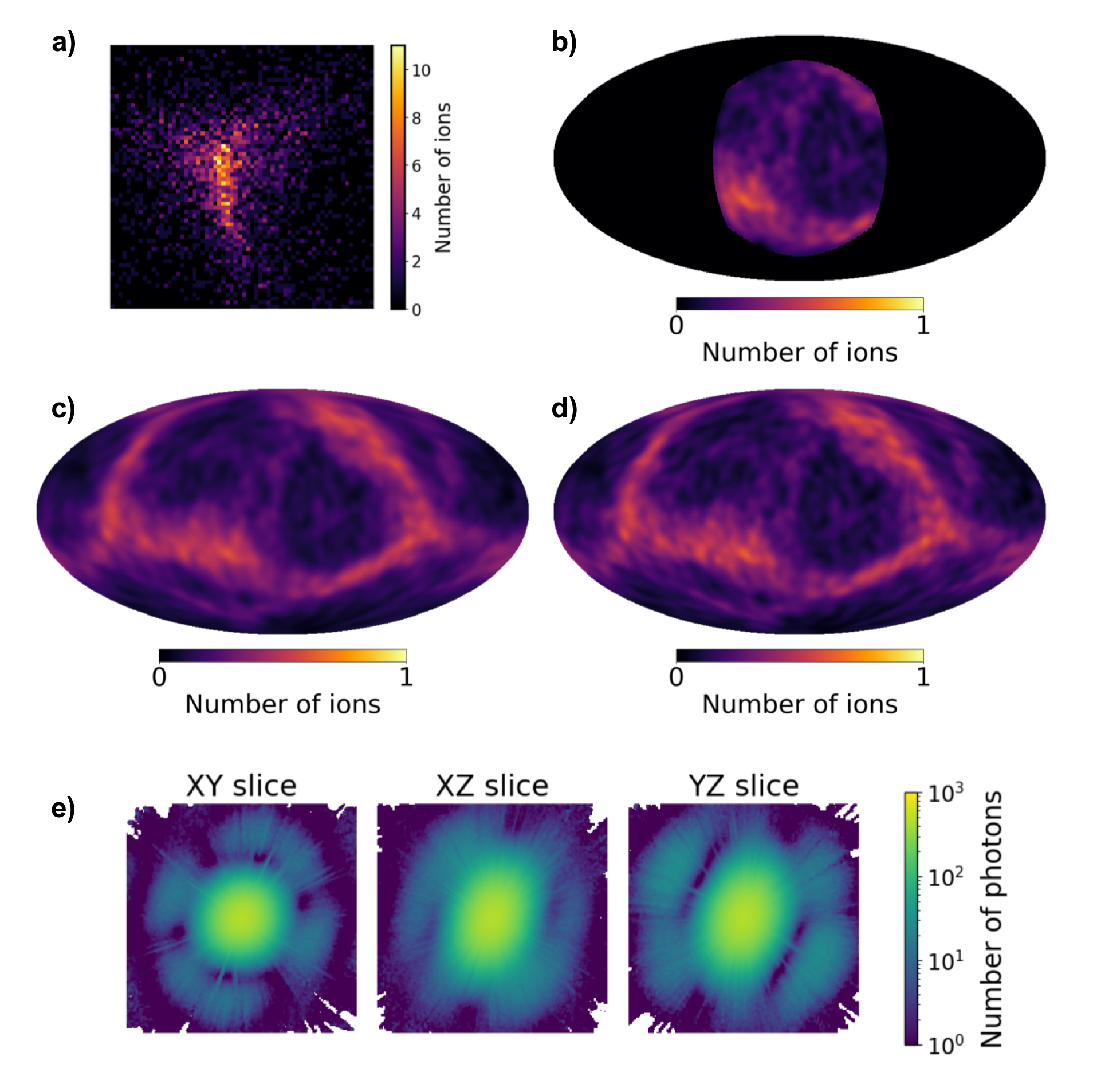
L’analogie qui rend cela tangible est simple. Prenez une orange, plantez-y cinquante cure-dents en tous sens, puis lâchez-la depuis une hauteur. Si vous retrouvez les cure-dents éparpillés au sol, leur disposition vous renseigne sur l’orientation qu’avait l’orange au moment de l’impact. Ici, les cure-dents sont des ions carbone, azote, oxygène — et le sol est un détecteur sphérique qui enregistre leur point d’arrivée.
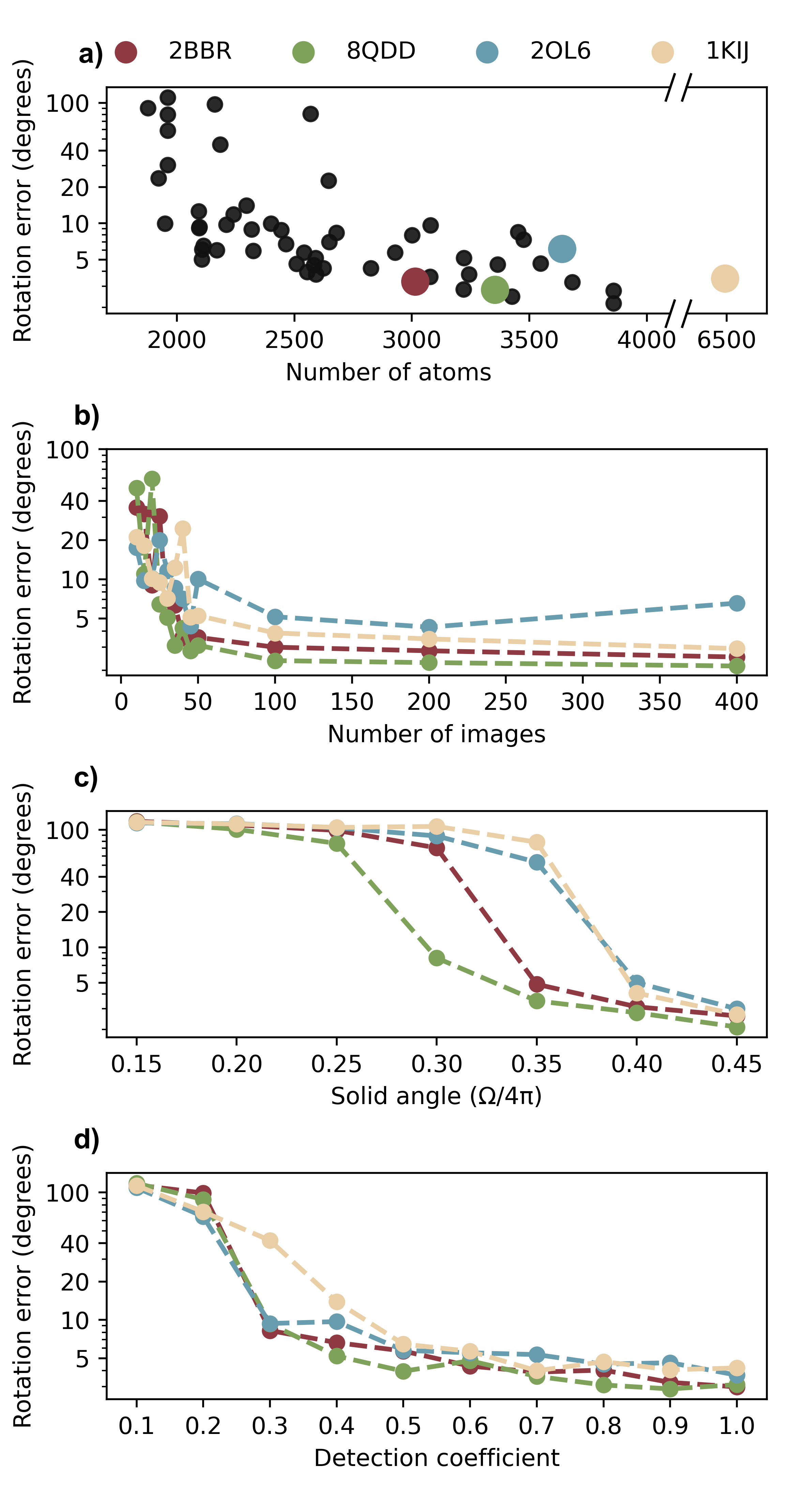
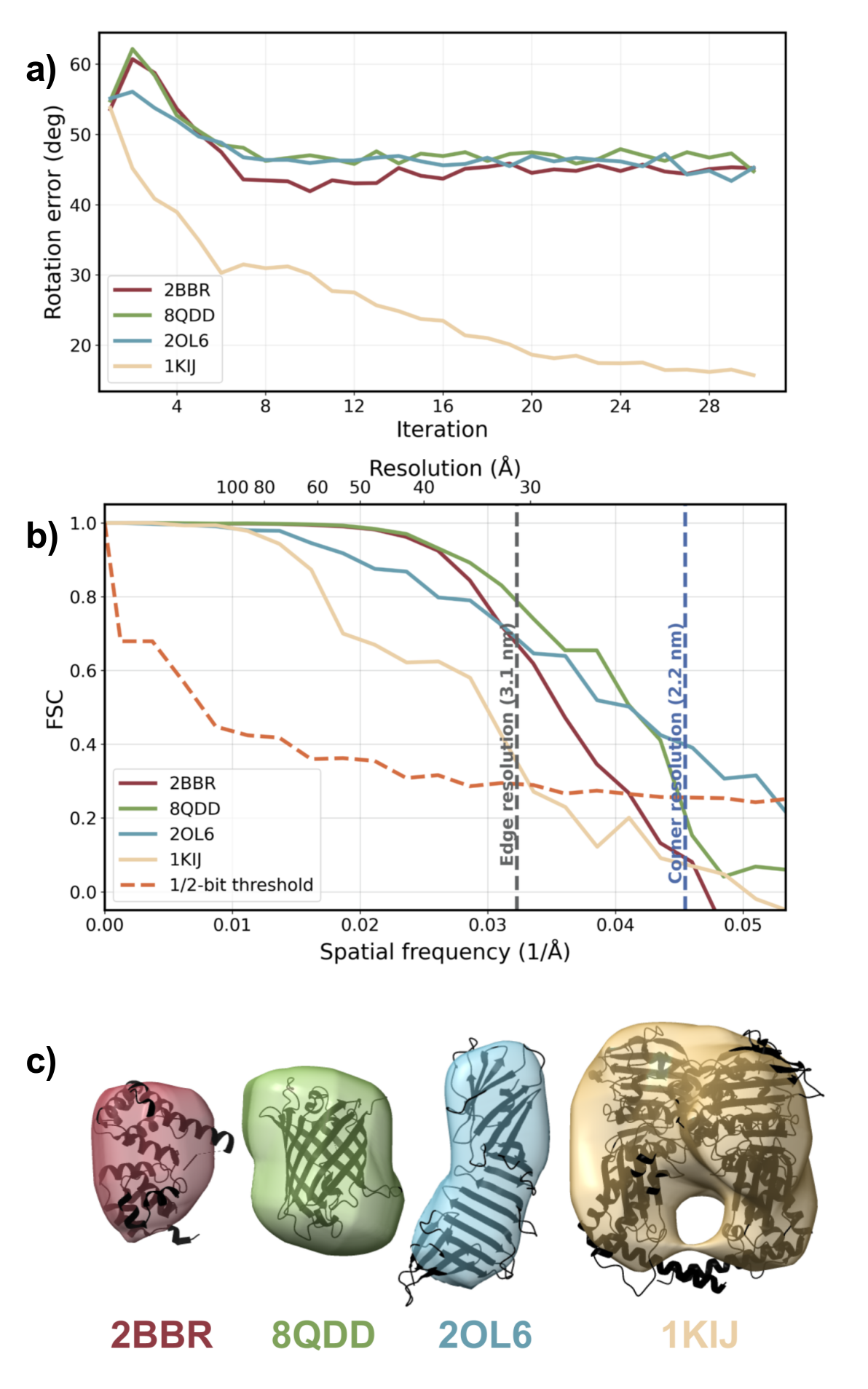
En projetant les positions des ions captés sur cette sphère, puis en recalant les empreintes les unes par rapport aux autres, l’algorithme retrouve l’orientation de la protéine à environ 5° près. Selon les auteurs, la méthode parvient à converger avec 50 à 100 mesures seulement là où l’approche classique par diffraction échoue avec 400 clichés — à condition, toutefois, de capter au moins un tiers de la sphère d’explosion. Précision importante : la méthode ne fonctionne pas en lieu et place de la diffraction, elle la complète. Les empreintes ioniques et les figures de diffraction X constituent deux sources d’information physiquement indépendantes, et c’est leur combinaison qui donne au système sa robustesse.
L’équipe a éprouvé l’approche sur 56 protéines simulées, de 1 800 à 6 500 atomes. Au-delà de 2 500 atomes, la convergence est stable et reproductible. En dessous, les trajectoires ioniques deviennent trop désordonnées : la molécule est trop petite pour que l’explosion porte une signature géométrique lisible. Une limite claire, que les auteurs ne cherchent pas à minimiser.
Il faut ici marquer une pause et rappeler ce que cette étude est, et ce qu’elle n’est pas encore. Tout cela reste numérique. Aucun faisceau réel n’a frappé une protéine dans ces conditions. Les simulations reproduisent les paramètres des grands lasers à électrons libres existants — notamment l’XFEL européen (Hambourg) et le LCLS (Stanford) — et la preuve de concept est entièrement algorithmique. La validation expérimentale reste à venir. C’est une étape que les auteurs signalent eux-mêmes, et que le lecteur doit garder en tête : entre une démonstration numérique convaincante et une technique d’imagerie opérationnelle, il y a souvent plusieurs années de travail acharné, d’obstacles imprévus, et parfois de déceptions.
Les enjeux, si la méthode tient ses promesses, sont considérables. De nombreuses protéines d’importance médicale — récepteurs membranaires, canaux ioniques, enzymes impliquées dans des maladies neurodégénératives — résistent à la cristallisation ou n’existent que dans des états transitoires, fugaces, inaccessibles aux méthodes classiques. Pouvoir déterminer leur structure, ou au moins leur orientation, à partir d’une seule molécule en liberté changerait la donne pour la conception de médicaments et la compréhension des mécanismes cellulaires.
Les auteurs ouvrent aussi une perspective encore plus spéculative, qu’il convient de signaler comme telle. Si chaque protéine produit une empreinte d’explosion suffisamment caractéristique, ils envisagent de pouvoir un jour remonter directement de la destruction à la structure — sans recourir à la diffraction du tout. Ce serait une imagerie moléculaire fondée exclusivement sur les débris, une lecture de la forme dans sa propre désintégration. L’hypothèse est séduisante, et les auteurs la qualifient eux-mêmes de hautement spéculative. Elle ouvrirait la voie à une imagerie structurale sans cristal ni synchrotron — mais c’est un horizon lointain, pas une promesse imminente.
Ce qui reste, en attendant, c’est une idée belle dans sa logique : que la destruction d’une chose peut en révéler la forme. Que l’explosion, loin d’être la fin de l’histoire, en est peut-être le chapitre le plus informatif.
Source
André T., Bellisario A., Timneanu N., Caleman C. et al. (Université d’Uppsala) — Orientation Reconstruction of Proteins using Coulomb Explosions — arXiv:2603.24553v1 (identifiant à vérifier sur le dépôt arXiv ; le format correspond aux soumissions de mars 2026).
À lire aussi sur Mémorabilité :
- Dans les pas d’un sanglier berlinois, ce que la géométrie d’un trajet révèle
- Persistance contextuelle dans un dépôt Git : le défi de la continuité pour les agents IA
- Tâches d’induction, d’analogie et de causalité : des écarts de performance marqués dans les grands modèles de langue
Figures originales du paper