
Vingt mille gènes s’expriment simultanément dans chacune de vos cellules. Vingt mille instruments qui jouent ensemble, sans chef d’orchestre visible, et dont la partition change à chaque seconde — quand un virus frappe, quand une hormone passe, quand un médicament inconnu se glisse entre les rouages. Or personne, jusqu’ici, ne savait écrire cette partition avant qu’elle ne soit jouée. Personne ne savait dire : si j’injecte telle molécule dans cette cellule, voici les gènes qui vont s’emballer, ceux qui vont se taire, et ceux qui ne bronceront pas. C’est pourtant ce que prétend faire Lingshu-Cell, un modèle génératif décrit dans une prépublication récente de Han Zhang et ses collègues (arXiv:2603.25240v1, non encore évaluée par les pairs au moment de la rédaction). Un double numérique de la cellule, capable de produire une réponse simulée à une perturbation qu’aucune cellule réelle n’a subie. La promesse est immense. La question est de savoir si elle tient.
Pour mesurer ce qui se joue, il faut d’abord toucher la matière première. Depuis une quinzaine d’années, la transcriptomique unicellulaire — le séquençage de l’ARN cellule par cellule, souvent désigné par le sigle ARN-seq unicellulaire (single-cell RNA-seq) — a transformé la biologie en science du dénombrement. On compte, littéralement, les molécules d’ARN présentes dans chaque cellule, gène par gène. Le résultat tient dans un tableau immense : chaque ligne une cellule, chaque colonne un gène, chaque case un nombre entier — zéro, un, trois, dix-sept copies. Des milliards de chiffres accumulés dans des bases publiques. Et dans ce déluge, une frustration lancinante : ces tableaux décrivent ce qui est, jamais ce qui pourrait être.
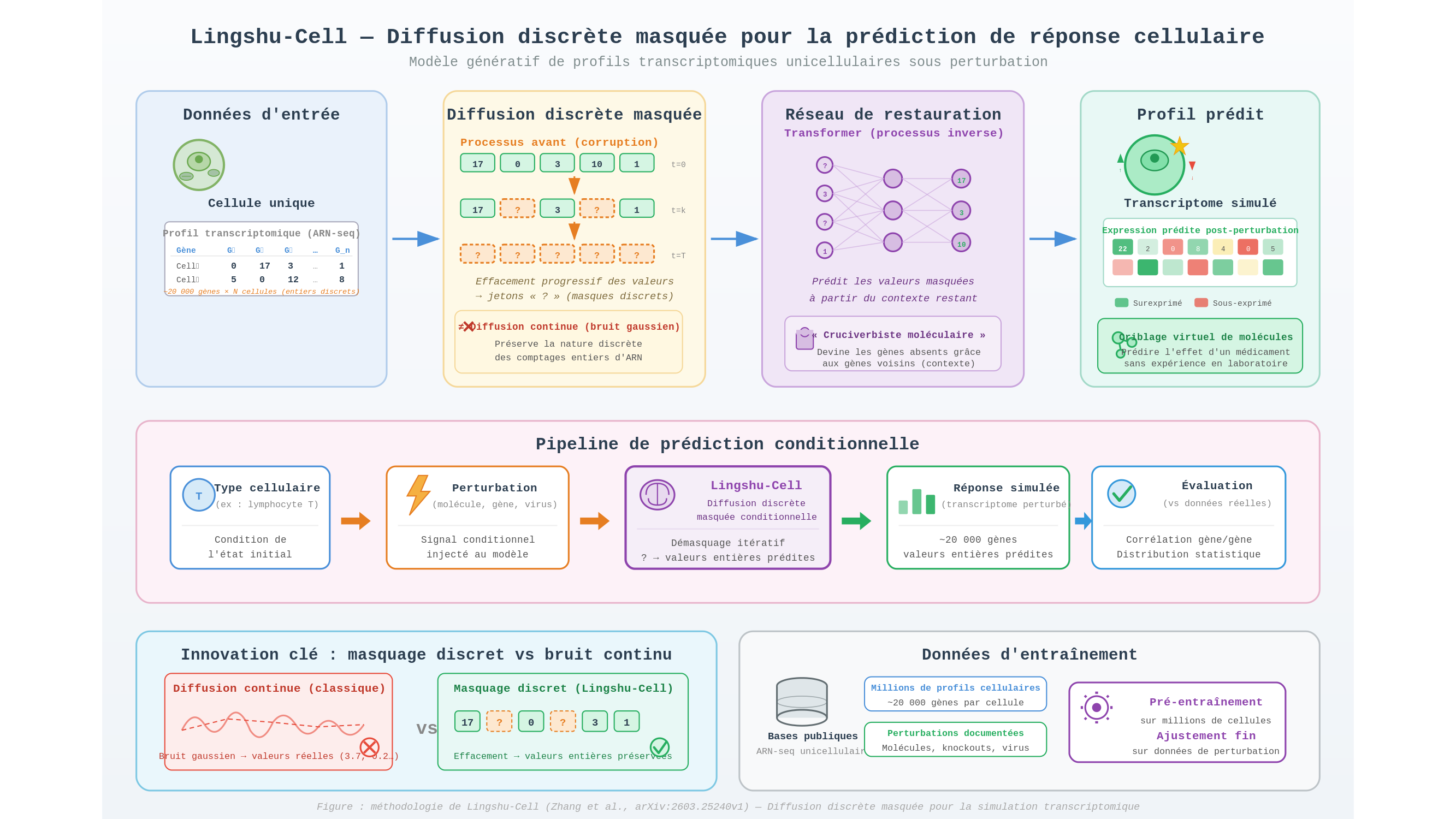
Plusieurs modèles fondateurs — Geneformer, scGPT — ont tenté de dompter cette masse en ajustant les paramètres de vastes réseaux de neurones sur des millions de profils. Leurs représentations permettent de classer, regrouper, annoter des états cellulaires avec efficacité. Elles produisent un instantané, pas un film. La différence avec Lingshu-Cell tient en un mot : génératif. Le modèle ne se contente pas d’étiqueter un état cellulaire ; il modélise la distribution statistique complète de ces états, et peut en engendrer de nouveaux — comme un romancier qui, ayant lu dix mille biographies, ne se contenterait plus de résumer des vies existantes mais en inventerait de plausibles, avec leurs bifurcations et leurs impasses.
Le mécanisme qui rend cela possible mérite qu’on s’y arrête, parce qu’il est d’une élégance inhabituelle. Les modèles de diffusion — ceux-là mêmes qui ont conquis la génération d’images — fonctionnent en deux temps : on corrompt les données en y ajoutant progressivement du bruit, puis on ajuste les paramètres d’un réseau pour inverser le processus, retrouver le signal à partir du chaos. Dans leur version classique, le bruit est gaussien, continu, lisse. Le problème, c’est que les comptages d’ARN ne sont pas lisses. Ce sont des entiers : zéro ou dix-sept, jamais 3,7. Appliquer une diffusion continue à ces données, c’est tenter de jouer du clavecin avec des moufles — on produit du son, mais on perd la finesse des touches. La diffusion discrète masquée (masked discrete diffusion) contourne l’obstacle avec un geste simple : au lieu de noyer les données dans un brouillard gaussien, elle efface progressivement des valeurs, les remplace par un jeton « inconnu ». Le réseau est alors optimisé pour restaurer ce qui manque en s’appuyant sur le contexte — un cruciverbiste moléculaire qui devine le mot absent à partir des lettres voisines. Le procédé respecte la nature discrète des comptages biologiques, et c’est ce qui fait sa force.
Reste à voir ce qu’on en fait. L’application visée par Zhang, Yuan, Yuan, Xu et Bian est la prédiction de réponse aux perturbations. Le scénario est le suivant : on fournit au modèle le profil transcriptomique d’une cellule au repos, on lui indique qu’un médicament X ou une mutation Y va être appliqué, et on lui demande de générer le profil résultant. La cellule virtuelle répond sans qu’aucune cellule réelle n’ait été sacrifiée. Pour un chimiste en quête de molécules thérapeutiques, la perspective est vertigineuse : cribler des milliers de composés in silico, éliminer les candidats inertes ou toxiques, ne valider en laboratoire que les survivants. L’économie potentielle — en temps, en argent, en vies animales — serait considérable.
Mais il faut ici résister à l’enthousiasme et poser les questions qui grattent.
La première est celle de la couverture. La transcriptomique unicellulaire ne capture qu’une couche de la réalité cellulaire. Elle ignore les protéines, l’épigénétique, l’organisation spatiale, les signaux mécaniques, les conversations entre cellules voisines. Simuler une cellule à partir de son seul transcriptome, c’est prédire l’humeur d’un quartier en ne lisant que le courrier de ses habitants : on saisit des tendances, on rate l’essentiel de la vie sociale. Les auteurs le concèdent implicitement en positionnant leur outil comme centré sur la transcriptomique, pas comme un simulateur cellulaire intégral. La nuance est capitale, et elle se perd vite dans l’élan des communiqués.
La seconde question est plus redoutable encore : celle de l’extrapolation. Les modèles génératifs excellent à interpoler, à produire des échantillons plausibles entre les exemples d’entraînement. Mais que se passe-t-il quand le médicament testé appartient à une famille chimique jamais vue ? Quand la mutation touche un gène rare, peu représenté dans les données ? La biologie, avec sa combinatoire explosive — vingt mille gènes, des centaines de types cellulaires, des milliers de perturbations possibles — est un terrain impitoyable pour les réseaux profonds qui s’aventurent hors de leur zone de confort. C’est précisément là, dans l’inconnu radical, que le pharmacologue a le plus besoin du modèle — et c’est là que le modèle est le plus fragile. À noter : la prépublication ne publie pas encore de résultats de comparaison quantitative avec les modèles existants, ce qui rend toute évaluation définitive prématurée.
Il serait pourtant injuste de n’y voir qu’une promesse supplémentaire dans un champ qui n’en manque pas. Le saut conceptuel est réel. Passer d’un modèle qui produit un portrait figé de la cellule à un modèle qui explore un paysage — l’ensemble des états accessibles, avec leurs probabilités — change la nature même des questions qu’on peut poser. Ce n’est plus « qu’est-ce que cette cellule ? », c’est « que deviendrait-elle si… ? ». Et cette capacité conditionnelle ouvre sur quelque chose de plus profond que la seule pharmacologie : en ajustant ses paramètres sur des millions de profils, le réseau encode des régularités transcriptomiques suffisamment fines pour produire des prédictions cohérentes. Pas une « compréhension » — le mot serait trompeur — mais une cartographie statistique de la grammaire du vivant. Jusqu’où ces régularités reflètent-elles de véritables mécanismes biologiques ? Ou ne sont-elles que des corrélations de surface, brillantes mais fragiles dès que le contexte dérape ?
La course aux « cellules virtuelles » est aujourd’hui l’un des fronts les plus encombrés de la biologie computationnelle. Consortiums internationaux, géants du numérique, laboratoires pharmaceutiques — tout le monde investit. Lingshu-Cell s’y inscrit avec une proposition technique soignée. Mais la distance entre un modèle qui génère des profils transcriptomiques plausibles et un outil qui transforme la découverte de médicaments se mesure en années de validation, en confrontations avec le réel, en échecs instructifs que la prépublication ne peut pas encore raconter.
Et c’est peut-être là que se cache le paradoxe le plus fécond de l’entreprise. Chaque prédiction ratée du modèle — chaque cellule réelle qui refuse de se comporter comme son double numérique — sera un indice. Le signe qu’un mécanisme inconnu est à l’œuvre, qu’un gène ignoré pèse plus qu’on ne le pensait, qu’une interaction manque à la carte. Les meilleurs miroirs sont ceux qui déforment juste assez pour qu’on aperçoive, dans la distorsion, ce qu’on n’avait jamais regardé.
À lire aussi sur Mémorabilité :
Sources
- Han Zhang, Guo-Hua Yuan, Chaohao Yuan, Tingyang Xu, Tian Bian, « Lingshu-Cell: a masked discrete diffusion model for transcriptomic state simulation and perturbation response prediction », prépublication arXiv, 2026. arXiv:2603.25240v1
- Christina V. Theodoris et al., « Transfer learning enables predictions in network biology », Nature, 2023. DOI:10.1038/s41586-023-06139-9 [Geneformer]
- Haotian Cui et al., « scGPT: toward building a foundation model for single-cell multi-omics using generative AI », Nature Methods, 2024. DOI:10.1038/s41592-024-02201-0


